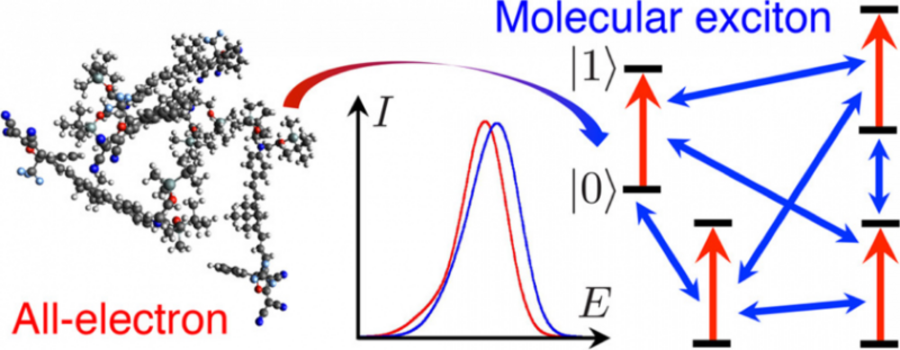
Aleksey's research comparing absorption spectra predicted by the exciton model and TDDFT is published in JCTC. In this paper, we examined the validity and accuracy of the exciton model for disordered aggregates of highly polar chromophores in close proximity (with high number density). Disordered chromophore aggregates have applications in a variety of organic materials, including photovoltaics and nonlinear optics, and having a method that can accurately predict the optical properties of these materials is key to their optimization. This work examined the accuracy of the exciton model for linear optical properties (absorption spectra) and in future work we will examine the nonlinear optical properties of interest for organic electro-optic materials.
The various approximations made in the exciton model are first outlined and then systematically examined for polar chromophore aggregates, which poses a challenging test of the model. We find that, despite the high number density and large transition dipole moments in this system, the computed values of the inter-chromophore couplings are in the realm of applicability of the exciton model. We then compare the absorption spectra computed with the exciton model to those computed with TDDFT (TD-B3LYP and TD-wB97X) and CIS. Using TeraChem, our spectral comparisons are performed on aggregates of up to 10 chromophores (~1200 atoms); these large-scale electronic structure calculations allow us to obtain size-dependent trends and to validate the model for bulk systems. We find good agreement between the exciton model and all the electronic structure methods, despite TD-B3LYP predicting many low-energy charge-transfer transitions at similar energies to excitonic transitions. Spectral agreement is improved when the electrostatic environment of the chromophores is taken into account with atomic point charges. We find a size-dependent error in the exciton model, but this size-dependent error decreases and begins leveling off when more than 6 chromophores are included in the aggregate, showing that the exciton model will be appropriate for predicting the optical properties of much larger chromophore aggregates. Most computational chemists do not have the ability to benchmark the model against an electronic structure method for larger chromophore aggregates in order to test how the model will behave as the system size increases, and this test thus provides insight into how well the model will behave for bulk systems.