Using both static and dynamic approaches (e.g. ensemble, Franck-Condon, cumulant), we are developing and validating ways to model the linear and nonlinear optical spectroscopy of molecules in complex condensed phase environments. We are applying these methods to model systems (generalized Born oscillators and Morse oscillators) as well as to molecules in different solvents and chromophores in proteins to better understand the limits of various approaches and gain physical insight into how the environment controls excited state processes.
For example, we recently developed an approach for modeling the band shapes of electronic absorption spectra of molecules in solution that combines both ensemble sampling of solute-solvent configurations and vibronic effects. For dyes in solution, capturing both inhomogeneous broadening of the solvent environment (which may be non-Gaussian for strong solute-solvent interactions) and the contribution to broadening from vibronic transitions is necessary for obtaining the correct spectral shape. Our approach treats all temperature effects classically through ensemble sampling of the ground state potential energy surface (including anharmonic regions of the PES), whereas the vibronic fine structure resulting from nuclear wave functions is treated at 0K through the Franck-Condon principle. These two approaches are then combined by dressing the vertical excitation energies with the Franck-Condon vibronic shape function.
The solvation environment around a solute has a large influence on molecular properties, but the extent of long-range solvation effects is unclear. It is thus not known how much solvent needs to be included to compute converged properties. We aim to (1) determine the amount of QM solvent necessary to compute converged solute properties in MM and PCM environments and (2) establish QM, QM/MM, and MM methods for accurate sampling of solute-solvent interactions in molecular dynamics.

Using an exciton model, we’ve developed a way to add organic chromophore excitonic interactions into the electro-optic effects through a two-state model for the hyperpolarizability. Now we are extending this model to 3D systems and testing it on electric field poled and un-poled chromophore aggregates with the goal of providing insight into the design of the next generation of organic electro-optic materials.

We want to move beyond understanding chromophore optical properties at the single molecule level and also understand them when they are coupled together in a thin film material. Absorption spectra are often quite different in dilute solution than in the aggregate, and our research aims to relate the configuration of multiple chromophores to the shifting and broadening of the computed and measured absorption spectra.

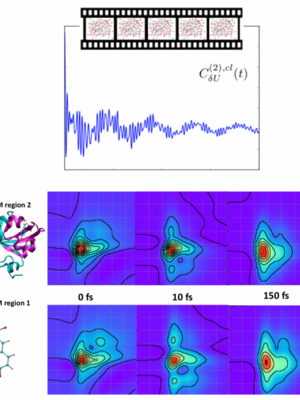
Electronic charge transfer is key to harnessing photon derived energy. After photo-excitation, the excited electrons must be transferred between phases, materials, or molecules. Thus, developing and benchmarking a method to directly model these electron dynamics is key to understanding these charge transfer processes at a fundamental level. Real-time TDDFT is a promising technique wherein the electron density matrix is explicitly propagated in time. This technique goes beyond the standard perturbative linear response TDDFT method, allowing the electron dynamics to respond to large perturbations such as an applied laser field. We use, develop, and validate this coherent superposition single-determinant density propagation method, examining the validity of the adiabatic approximation in the realm of strong-field perturbation of the electron density.