Makenzie's paper published in J. Phys. Chem. B shows how to more accurately model processes (such as electronic excitation) in solution by combining a quantum mechanical treatment of solvent molecules near to the active site with a polarizable continuum model.
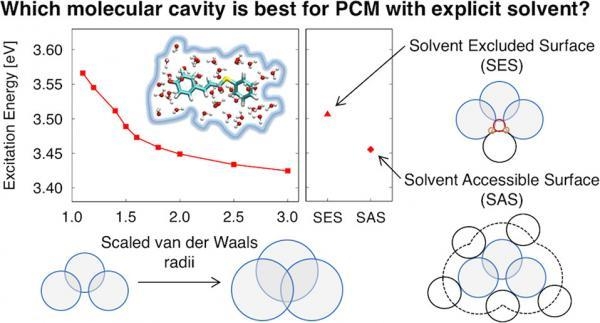
An accurate model should include some quantum mechanical (QM) solvent around the solute (to capture short-range effects such as charge transfer and hydrogen-bonding) and a classical model of the surrounding solvent (to capture long-range polarization effects). The polarizable continuum model (PCM) is a commonly used classical solvation model; however, using this model with explicit QM solvent presents a problem that has not been widely recognized. The choice of the solvation cavity is very important for obtaining physically correct results since unphysical double counting of solvation effects (both the QM solvent and the classical dielectric) can occur if a poor choice of cavity is used (for example, the default cavity in Gaussian09). We showcase this unphysical result in the context of excitation energies, and then show that the cavity can be corrected using a solvent excluded surface or a scaled van der Waals surface. Here, we compare the effects of the molecular cavity and self-consistent reaction field (SCRF) method on electronic excitation energies of three aqueous solutes using time-dependent density functional theory (TDDFT) within a combined QM/PCM approach. Previous studies show that QM properties are dependent on the choice of molecular cavity and SCRF method, but these studies typically do not include explicit QM solvent.